 Allgemeine Informationen
Allgemeine Informationen General
information
General
informationPatient*innenregister zur Schwerhörigkeit
Was ist ein Patientenregister?
Patient*innenregister unterstützen die Beteiligung von Patient*innen an der Forschung, die ansonsten möglicherweise Schwierigkeiten haben, an Studien zu seltenen Krankheiten teilzunehmen. Sie werden von Forscher*innen entwickelt, die Expert*innen für eine bestimmte Krankheit sind, um mit Hilfe der Patient*innen Informationen zu sammeln, die zum besseren Verständnis der Krankheit beitragen können. Patient*innenregister unterstützen die Erforschung von Krankheitsursachen und Therapie sowie Studien zum Krankheitsverlauf und können der Rekrutierung von Teilnehmer*innen für klinische Studien zur Erprobung neuer Therapien dienen.
Mit Hilfe der Otoferlin- und CABP2-Register möchten wir die genetischen Mutationen in den OTOF- und CABP2-Genen und deren Auswirkungen auf das Gehör im Laufe des Lebens verstehen, auch in Bezug auf Hörrehabilitation (Hörgeräte, Cochlea-Implantate, zukünftige Gentherapie) und die Auswirkungen von Veränderungen der Körpertemperatur (bei einigen Otoferlin-Mutationen wurde ein temperaturabhängiger Hörverlust beobachtet), um unsere Forschung zu den genetischen Ursachen der Schwerhörigkeit weiter voranzutreiben.
Wie häufig ist Taubheit? Welche Arten von Gehörlosigkeit gibt es?
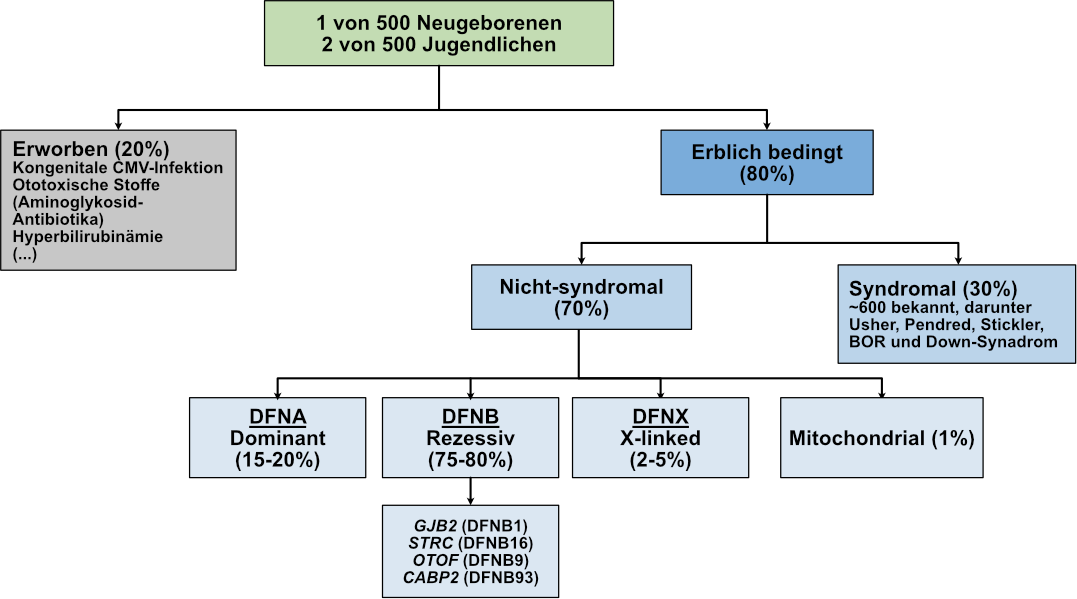
In vielen Teilen der Welt wird im Rahmen des allgemeinen Neugeborenen-Hörscreenings mit Untersuchungen auf Hörverlust begonnen. Aus diesen Daten wissen wir bereits, dass bei etwa einem von 500 Neugeborenen eine behandlungsbedürftige Schwerhörigkeit vorliegt und dass sich diese Zahl im Jugendalter etwa verdoppelt und im Laufe des Lebens stetig weiter ansteigt (Abbildung 1). Hierbei ist zu beachten, dass Schwerhörigkeit tendenziell unterdiagnostiziert ist und dass es manchmal viele Jahre dauern kann, bis eine Hörminderung klinisch festgestellt wird. Daher ist es auch bei Verdacht auf Schwerhörigkeit sehr wichtig, dass Audiolog*innen und HNO-Ärzt*innen das Hörvermögen genau untersuchen und testen.
Wir können Hörverlust und Taubheit anhand verschiedener Parameter beschreiben. Einer dieser Parameter ist, ob sie auf genetische oder umweltbedingte Ursachen zurückzuführen sind. Bei früh einsetzender Schwerhörigkeit geht man davon aus, dass sie überwiegend genetisch bedingt ist. Von den vererbten Formen der Schwerhörigkeit treten 70 % als isolierte Schwerhörigkeit auf, die auch als nicht-syndromale Schwerhörigkeit bezeichnet wird, während etwa 30 % der erblich bedingten Fälle als syndromale Schwerhörigkeit mit zusätzlichen Merkmalen auftreten, die andere Organsysteme betreffen. Von den isolierten Formen der Schwerhörigkeit treten 75-80 % als autosomal rezessive Form auf, was bedeutet, dass sowohl die von Mutter als auch die vom Vater vererbte Kopien eines Gens jeweils eine Mutation aufweisen, die an das Kind weitergegeben wird. Das Besondere an autosomal rezessiven Formen der Schwerhörigkeit ist, dass es normalerweise keine familiäre Vorgeschichte gibt und die Eltern in der Regel normal hören. Mutationen in den Genen GJB2 (DFNB1) und STRC (DFNB16) sind die häufigsten genetischen Ursachen für früh einsetzende Schwerhörigkeit. OTOF (DFNB9) ist ebenfalls ein wichtiges Gen, das bei erblicher Schwerhörigkeit eine Rolle spielt und ein Ziel für potenzielle Gentherapien darstellt (Abbildung 1). Schwerhörigkeit kann auch im Rahmen eines autosomal-dominanten Erbgangs vererbt werden, bei dem Mutationen in einer einzigen Kopie des betroffenen Gens Schwerhörigkeit verursachen können. In diesem Fall gibt es in der Regel eine nachvollziehbare Familiengeschichte mit mindestens einem betroffenen Elternteil und einem ähnlichen Verlauf der Schwerhörigkeit (z. B. Beginn und Schweregrad). Man geht davon aus, dass dies bei 15-20 % der früh einsetzenden Fälle von Schwerhörigkeit der Fall ist. Zu den selteneren Formen der erblichen Schwerhörigkeit gehören die X-chromosomal bedingte Schwerhörigkeit, die hauptsächlich Männer aufgrund von Mutationen in Genen auf dem X-Chromosom (2-5 %) betrifft, und die mitochondrial bedingte Schwerhörigkeit (~1 %) (Abbildung 1).
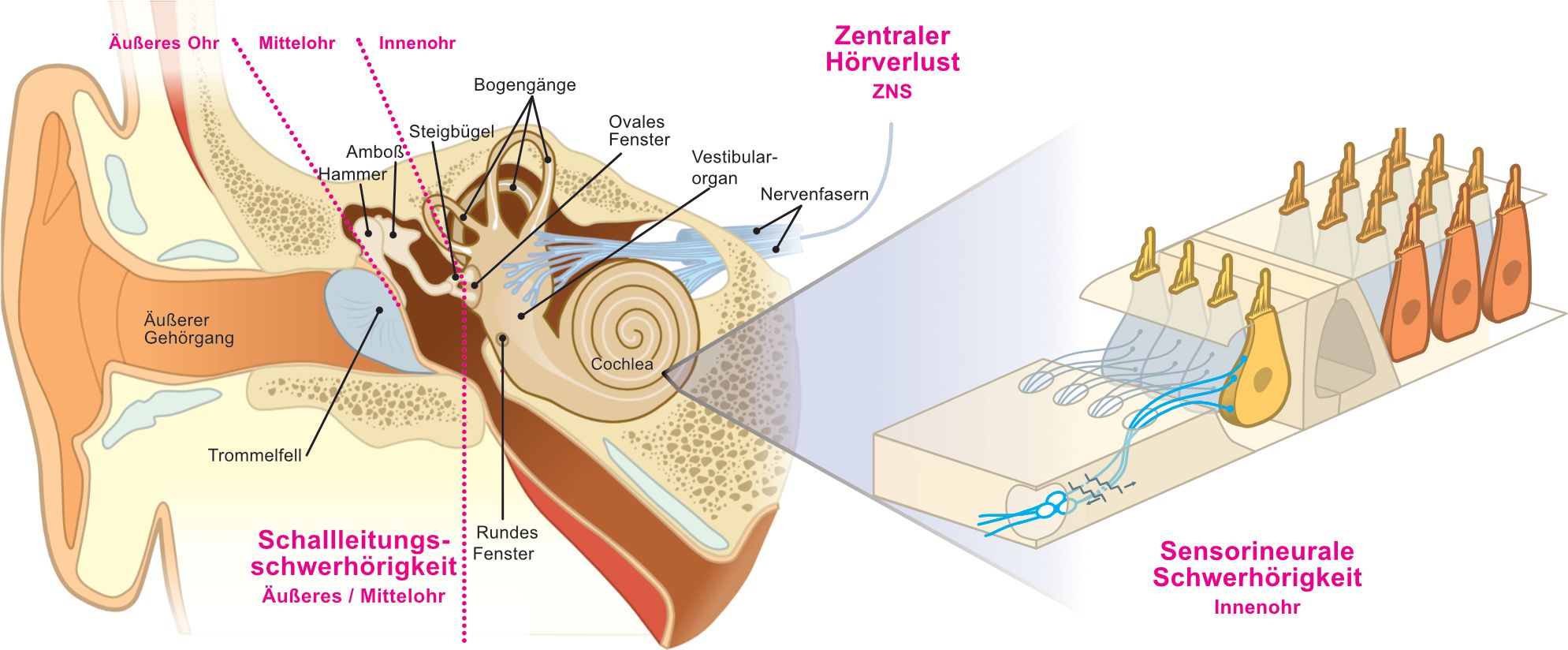
Schwerhörigkeit kann auch anhand des genauen Orts des Defekts im auditorischen System beschrieben werden. So kann eine Schädigung des Außen- oder Mittelohrs eine Schallleitungsschwerhörigkeit verursachen, während eine Schädigung der empfindlichen Zellen der Cochlea und des Hörnervs zu einer sensorinalen Schwerhörigkeit führen kann. Defekte an der Synapse der sensorischen Rezeptorzellen des Innenohrs, der so genannten Haarzellen, führen zu Synaptopathie, also der fehlerhaften Übertragung von Informationen auf die nachfolgenden Spiralganglionneurone des Hörnervs. Schäden an den Spiralganglionneuronen hingegen verursachen eine Neuropathie, also eine fehlerhafte Weiterleitung der Signale im Hörnerv. Eine Kombination von Schäden am Außen-, Mittel- und Innenohr kann zu einem kombinierten Hörverlust führen. Schließlich führt eine Schädigung der zentralen Hörbahn oder des Hörzentrums im Gehirn zu einem zentralen Hörverlust (Abbildung 2).
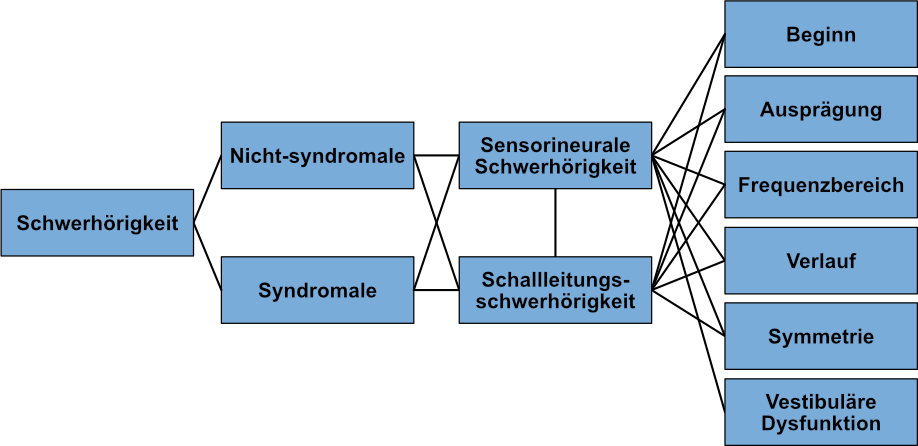
Molekulargenetische Untersuchungen können nicht nur Aufschluss darüber geben, welche Zellen im auditorischen System als Ursache für Hörverlust in Frage kommen, sondern auch darüber, welche Proteine betroffen sind und welche zellulären Funktionen betroffen sein könnten. Dies kann zu einem besseren genbasierten Verständnis anderer Merkmale im Zusammenhang mit dem Auftreten von Hörverlust führen, wie z. B. dem Schweregrad und welche Frequenzen betroffen sein werden, ob sich der Hörverlust im Laufe der Zeit verändert oder fortschreitet, ob beide Ohren gleichermaßen betroffen sein werden und ob eine vestibuläre Dysfunktion, also eine Gleichgewichtstörung, auch vorliegen könnte (Abbildung 3).
Wie kann Schwerhörigkeit behandelt werden?
Die Behandlung von Schwerhörigkeit erfolgt in enger Zusammenarbeit von HNO-Ärzt*innen und Audiolog*innen und wird nach einer eindeutigen Diagnose festgelegt. Hörgeräte, die die fehlenden Frequenzen verstärken, sind oft hilfreich für Personen mit einer ausreichenden Rest-Hörfunktion. Für Personen mit einem schwereren oder hochgradigen Hörverlust kommen häufig für Cochlea-Implantate in Frage, die den Hörnerv elektrisch stimulieren und dabei die geschädigte Stelle im Innenohr umgehen. Dies sind nach wie vor die wirksamsten Mittel zur Behandlung von Hörverlusten. Wichtige Forschungsarbeiten werden fortgesetzt, um diese Behandlungsmöglichkeiten weiter zu verbessern (siehe auch „Hören mit Licht“).
In präklinischen Studien haben sich gezielte Gentherapien als vielversprechende Möglichkeiten herausgestellt, in Zukunft die genaue genetische Ursache für Hörverlust zu beheben. Diese Therapien sind jedoch noch nicht zur Anwendung im Menschen zugelassen. Wir haben wichtige Beiträge zum Verständnis der Otoferlin-bedingten Taubheit geleistet und treiben die präklinische Arbeit an solchen Therapien weiter voran. Wir möchten diese Gelegenheit nutzen, um das Register für die Einbeziehung von Otoferlin-Patient*innen in die Forschung zu nutzen. Dadurch möchten wir weitere Erkenntnisse über die Otoferlin-bedingte Taubheit gewinnen und zukünftige Gentherapien vorbereiten.
Weitere Informationen
Weitere Informationen können Sie in diesem Übersichtsartikel nachlesen (aus der Zeitschrift HNO).